
Mitochondrial Dysfunction and Immune Suppression
Mutations that negatively impact mitochondrial function are highly prevalent in humans and lead to disorders with a wide spectrum of disease phenotypes, including deficiencies in immune cell development and/or function. Yet uncovering the molecular underpinnings of systemic signaling circuits crucial to the integrity of immune system function remains a major barrier to understanding the etiology of many of these metabolic disorders.
We previously used a suite of transgenic mice with localized loss of cytochrome c oxidase (COX) activity to identify a copper-linked, systemic signaling circuit activated by disruption of mitochondrial function in the murine liver or heart that results in atrophy of the spleen and thymus and causes a peripheral white blood cell deficiency (Jett et al., 2023). We further demonstrated that the white blood cell deficiency is caused by hepatic secretion of α-fetoprotein, which requires copper and the cell surface receptor CCR5 to promote cell death (Jett et al., 2023).
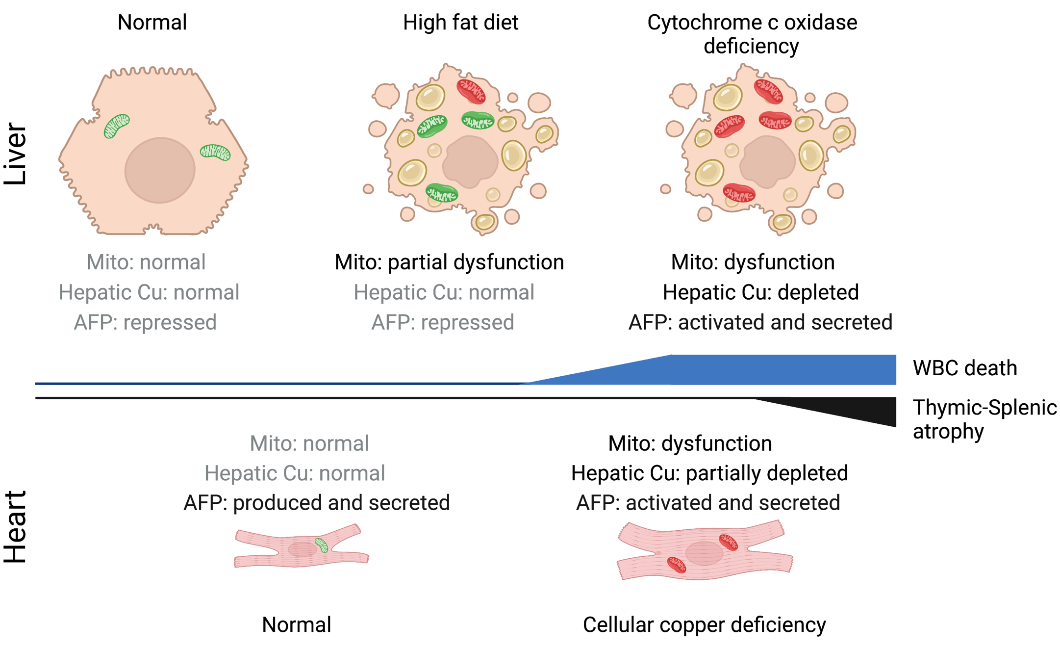
Our on-going collaboration with the Pioli lab seeks to expand upon these foundational observations to further understand the consequences of tissue-specific mitochondrial deficits to the integrity of immune system function. Recently, we demonstrated that mice lacking the essential COX assembly factor SCO1 in their hepatocytes exhibit metabolic defects extrinsic to the hematopoietic compartment that lead to a pan-lymphopenia represented by the severe loss of both B and T cells (Pioli et al., 2024). We also showed that immune defects in these mice are associated with the loss of bone marrow lymphoid progenitors common to both lineages, and early signs of autoantibody-mediated autoimmunity. Our findings collectively point to a significant role for hepatocyte dysfunction as an instigator of immunodeficiency in patients with congenital mitochondrial defects who suffer from chronic or recurrent infections. As such, we continue to characterize this novel systemic signaling circuit because we believe it will greatly advance our understanding of physiology, and lead to the identification of therapeutic targets that can be leveraged to improve the quality of life of some mitochondrial disease patient cohorts.